
「どの試験デザインが自分たちの研究に最適かわからない」。
多くの医療従事者や開発担当者が、この壁に直面します。
適切なデザインなしに、信頼できるデータは生まれません。
本記事では、ランダム化比較試験(RCT:参加者をランダムに割り付けて治療効果を比較する試験)などの基礎から、近年注目されるバスケット試験・アダプティブデザイン・ベイズ流といった革新的手法(CID:従来型を超えた複雑で新しい臨床試験デザイン)までを体系的に解説します。
独立行政法人医薬品医療機器総合機構(PMDA)の「臨床試験の一般指針」に基づき、科学的妥当性と倫理性を両立するための知識を網羅しました。
治験に関わる全てのプロフェッショナルのための「持ち運べる教科書」として活用してください。

弊社は医療の未来を支える臨床研究の支援を通じ、社会に貢献することを理念として掲げております。
ご相談いただく研究内容や条件に応じて、柔軟かつ誠実に対応させていただきます。
臨床研究の実施にあたりご支援が必要な場合は、ぜひお気軽にお問い合わせください。
臨床試験デザインとは? 目的と「3つの基本要素」
なぜ「デザイン」が最重要なのか
臨床試験におけるデザインは、建物の「設計図」に相当します。
最も重要な役割はエビデンスレベルの担保です(エビデンスレベル:研究結果の信頼性の強さを示す概念)。
不適切な試験デザインで収集されたデータは、どれほど高度な統計解析を行っても信頼できる結論を導き出せません(GIGO: Garbage In, Garbage Out:入力データの質が悪ければ結果も悪くなるという考え方)。
PMDAの「臨床試験の一般指針」においても、「2.2 科学的な臨床試験のデザインと解析」として、バイアスを排除し、事前に計画された手順に従うことの重要性が強調されています(バイアス:結果を歪める系統的な偏り)。
また、デザインを決定する際は、PICO(Patient: 対象、Intervention: 介入、Comparator: 比較対照、Outcome: 結果:研究課題を整理する基本枠組み)の関係性を明確にすることが第一歩となります。
デザインを構成する3つの柱
臨床試験の科学的妥当性を担保するために不可欠な、3つの基本要素があります。
対照群の設定 (Control):
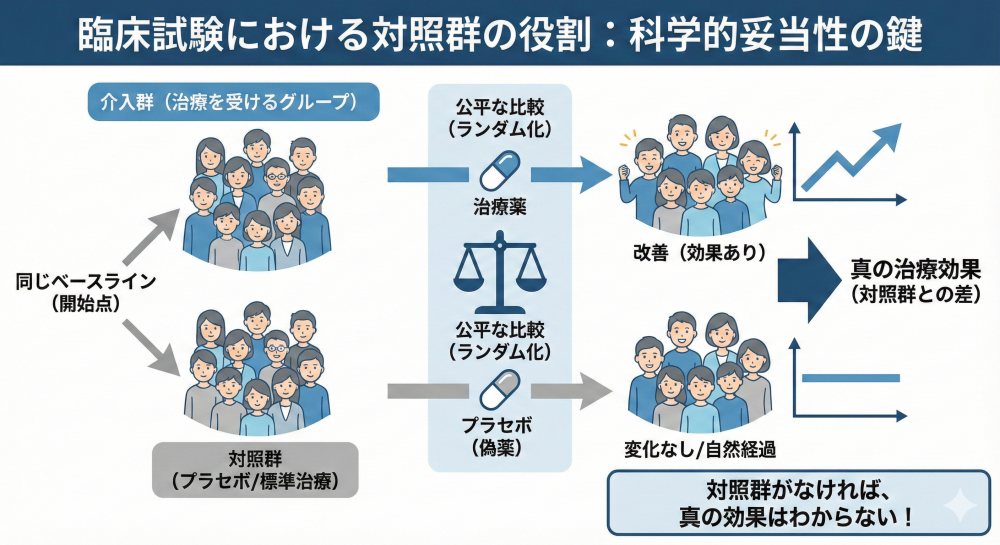
新薬の効果を正しく評価するには、比較対象が必要です。
プラセボ対照(Placebo:有効成分を含まない偽薬)、実薬対照(既存の標準治療)、無治療対照があります。
希少疾患などで対照群を置くことが倫理的・物理的に困難な場合に限り、過去のデータを用いるヒストリカルコントロール(外部対照)(Historical Control:過去や外部のデータを比較対象として用いる方法)が許容されるケースがあります。
ランダム化 (Randomization):
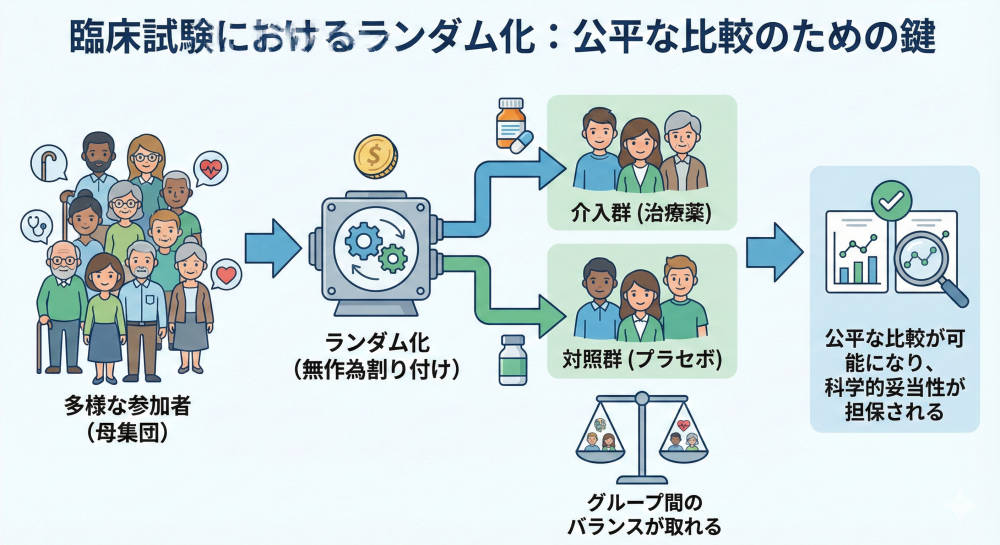
参加者を作為なく各群に割り付ける操作です。単純ランダム化、ブロックランダム化、層別ランダム化などの手法があります。
最大の目的は、年齢や重症度といった既知の因子だけでなく、「未知の交絡因子」を群間で均等にし、バイアスを制御することにあります(交絡因子:介入と結果の関係を見えにくくする別の要因)。
盲検化 (Blinding):
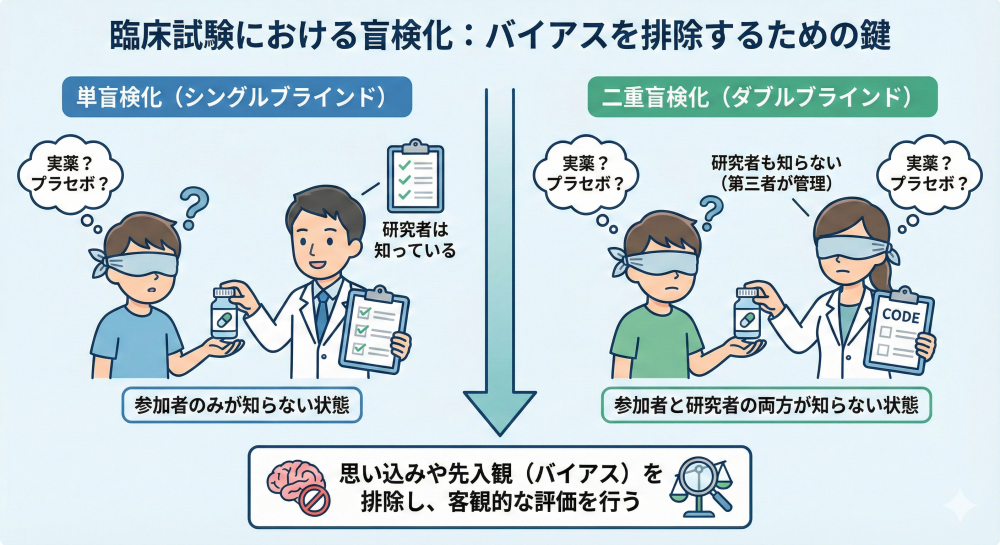
試験に参加する関係者が割り付けられた治療内容(例:実薬かプラセボか)を知らない状態にすることで、予断や期待によるバイアスを防ぐ手法です。
単盲検(参加者のみ隠す)、二重盲検(参加者と医師共に隠す)、オープンラベル(隠さない)に分類されます。
近年では、治療はオープンラベルで行いつつ、評価者のみを盲検化するPROBE法(前向きランダム化オープンラベル評価者盲検:評価の公平性を高める工夫)も活用されています。
代表的な試験デザインの分類と特徴
ここでは試験の構造による分類を解説します。
また、イメージしやすいようにデザインごとの具体例も紹介します。
※あくまでも一例であり、実在する試験を指すものではありません
並行群間比較試験 (Parallel Group Design)
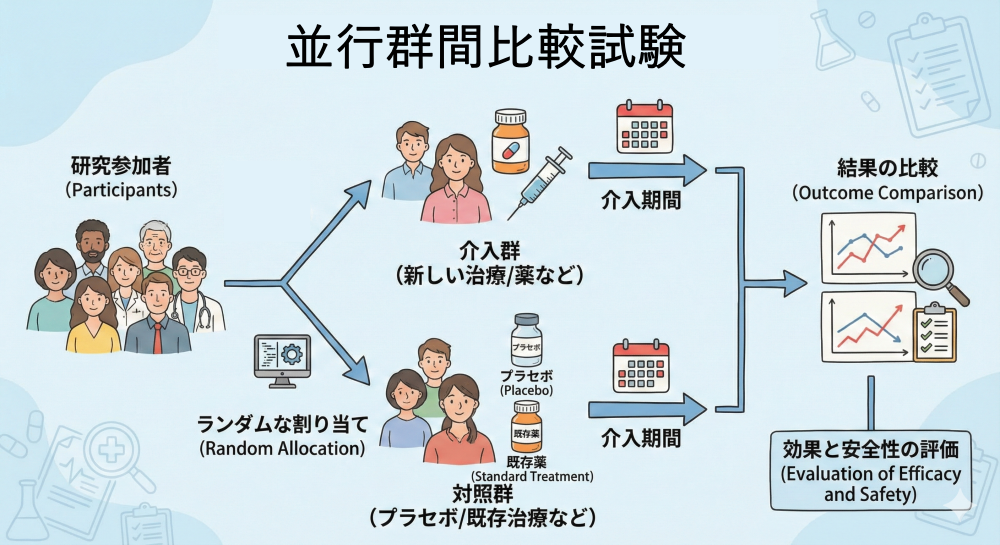
特徴: 臨床試験のゴールドスタンダード(最も標準的で信頼性が高いとされる方法)とされ、解析がシンプルで結果の解釈が容易です。
注意点: 群間の背景因子を揃えるために、比較的多くの症例数(サンプルサイズ $N$:解析に必要な参加者数)が必要となります。
例:花粉症の新しい飲み薬Aを試したい
200人の花粉症患者をランダムに2組に分ける。
・A群(100人):新薬Aを毎日
・B群(100人):標準薬Bを毎日
一定期間後にくしゃみ回数、鼻づまりスコア、副作用を比較。
クロスオーバー試験 (Crossover Design)
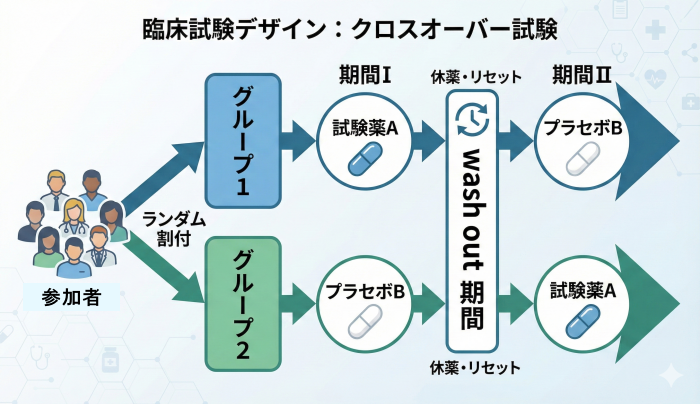
構造: 一人の参加者が、時期をずらして「実薬」と「プラセボ(または対照薬)」の両方を順に試します(自己対照:同じ人が自分自身の比較対象になる考え方)。
特徴: 個人差の影響を除外できるため、少人数でも高い検出力(検出力:本当に差があるときに差を見つけられる確率)を持ちます。
必須条件:
- 症状が安定している慢性疾患であること。
- 前の薬の影響を消すためのウォッシュアウト期間(ウォッシュアウト期間:前治療の影響が残らないように空ける期間)の設定。
- 前の治療効果が後の期間に影響しないこと(持ち越し効果がないこと)。
例:慢性の不眠症で、新しい睡眠薬Aが本当に効くか見たい
同じ患者がAとBの両方を体験する。
- 期間1(2週間):睡眠薬A
- 休薬(1週間):ウォッシュアウト
- 期間2(2週間):プラセボB
別のグループは順番が逆(B→A)。
各期間で 入眠までの時間や中途覚醒回数を比較。
その他の重要なデザイン
要因配置試験 (Factorial Design)
2種類の薬剤の単独効果と併用効果を同時に検証する場合などに用いられます(複数介入を効率よく評価する枠組み)。
例:高血圧予防で「減塩」と「運動」の効果を同時に検証したい
参加者を下記の4群にランダム化:
- 減塩あり + 運動あり
- 減塩あり + 運動なし
- 減塩なし + 運動あり
- 減塩なし + 運動なし
一定期間後に、収縮期血圧や拡張期血圧を比較。
群ランダム化試験 (Cluster RCT)
個人単位ではなく、「病院」や「地域」単位で割り付けを行います(集団単位で介入の効果をみる方法)。
例:学校単位で感染対策が効くかを見たい
A校:手洗い教育+教室に消毒ステーション追加
B校:通常の衛生指導
個人ではなく学校という“集団”をランダムに割り付けて、冬のインフルエンザ欠席率を比較。
必要なサンプルサイズが大幅に増える:同じ学校の生徒同士は生活環境が似ているため、データとしても似た傾向を持ちます(独立性が低い)。
そのため、統計的な「情報の価値」が目減りします。個人単位の試験と同じ感覚で人数計算をすると、本当は効果があるのに「差なし」という結果になりがちです。
同意取得と割り付けの「順序」を間違える 「A校は新しい対策あり」と決まった “後” に生徒へ参加を募ると、「新しい対策があるなら参加したい」という人が増え、群間の公平性が崩れてしまいます(選択バイアス)。
「全員の参加同意を取る → その後に学校を割り付ける」 という順序を厳守する必要があります。
マッチドペア試験
性別・年齢・重症度などの特徴が似た患者をペアにし、それぞれを別々の群に割り付けることで背景因子を調整する手法です(背景差をできるだけ小さくする工夫)。
例:術後の痛み止めAとBを比べたいが、患者の差を極力減らしたい
- 年齢
- 性別
- 手術内容
- 痛みの重症度
が似ている患者を2人1組でペアにする。
ペア内で
- 片方:鎮痛薬A
- 片方:鎮痛薬B
を使い、24時間の痛みスコアを比較。
開発フェーズごとの標準的デザインとがん領域の特例

第I相試験(探索的試験):安全性をどう見極めるか
SADとMAD
一般的な薬剤では、健常人を対象に単回投与(SAD:Single Ascending Dose:単回投与で用量を段階的に増やす試験)から開始し、安全性を確認後に反復投与(MAD:Multiple Ascending Dose:複数回投与で用量を増やす試験)へ移行します。
がん領域の第I相(Oncology Phase 1)
抗がん剤は細胞毒性が強いため、患者を対象に行われます。
3+3法
3人のコホート(Cohort:同じ用量など同条件の患者集団)ごとに用量を上げ、毒性(DLT:Dose Limiting Toxicity:用量制限毒性)の発現状況で次の用量を決める伝統的なアルゴリズムです。
例えば、新しい薬を「レベル1(少量)」から「レベル3(多量)」まで試すとします。
まず3人の患者さんに投与しました。
結果: 3人ともDLT(重い副作用)は出ませんでした(0/3人)。
判定: 「この量は安全」と判断し、次のレベルへ増量します。
次の3人に、倍の量を投与しました。
結果: 1人にDLTが出ました(1/3人)。
判定: 「たまたまこの人の体調が悪かったのか、薬が強すぎるのか?」判断がつきません。 そのため、同じレベル2で、さらに「3人」を追加します(これが”3+3″の名前の由来です)。
追加結果: 合計6人(最初の3人+追加の3人)で見ると、DLTが出たのは最初の1人だけでした(1/6人)。
最終判定: 「6人中1人なら許容範囲」として、次のレベルへ増量します。
次の3人にさらに多い量を投与しました。
結果: 2人にDLTが出ました(2/3人)。
判定: 「これは危険すぎる」と判断し、ここでストップします。
CRM (Continual Reassessment Method)
統計モデルを用いて、より正確にMTD(最大耐量:副作用が許容できる範囲での最大用量)を推定する手法です。
CRMは3+3法よりもさらに「数学的(統計学的)」に、最適な投与量を探り当てる方法です。
最初に、医師と統計家が「おそらくこの量で副作用〇〇%くらいになるだろう」という予測カーブ(曲線)を描きます。
最初の患者さんに投与します。
患者さんに副作用が出なかった場合 → コンピュータが計算:「思っていたより薬が弱い(安全)ようです。予測カーブを修正します。次はもっと多い量で30%を狙いましょう」
患者さんに副作用が出た場合 → コンピュータが計算:「思っていたより薬が強い(危険)ようです。予測カーブを修正します。次は少し量を減らして30%を狙いましょう」
これを患者さん1人(または数人)ごとに繰り返し、最終的に「副作用発生率がちょうど〇〇%になる量(MTD)」をピンポイントで特定します。
推奨用量決定後に症例を追加し、有効性のシグナルを早期に掴む工夫が行われます(安全性だけでなく効果の兆しも確認する目的)。
第II相試験(新薬としての「有望さ」を確認する段階):POCの確認
用量反応性の検討およびPOC(Proof of Concept:概念実証)の確認を行います。
簡単に言うと、POCは「机上の空論ではないことの証明」を意味します。
実際に少数の患者さんに投与し、「本当に薬として期待できるか」という確かな手応えを掴むことが、この試験の最大のゴールです。
単群試験の可否
予後不良ながんや希少疾患において、過去の患者データと比較して明らかに高い奏効率(腫瘍が明らかに縮んだ人の割合)が期待できる場合、対照群を置かない単群試験が行われることがあります(倫理的・実務的制約への対応)。
Simonの2段階デザイン
第一段階で効果が見られない場合、無益な治療を早期に中止(Futility stop:有効性が見込めないと判断して中止すること)し、患者の不利益と開発コストを抑えるデザインです。
主にがんの第II相試験で使われる、非常に有名な試験デザインです。
一言で言うと、「見込みのない薬は、早めに見切りをつけて試験をストップする(無効中止)」ための仕組みです。
第III相試験(検証的試験):優越性・非劣性・同等性
大規模なRCTにより、標準治療に対する位置づけを確定させます。
優越性試験
新薬が対照薬より優れていることを示す。
非劣性試験
新薬が対照薬より「劣っていない」ことを示す(効果が同程度でも安全性や利便性で価値がある場合に重要)。
これには厳密な「非劣性マージン」(Non-inferiority Margin:許容できる差の上限)の設定と、必要なサンプルサイズの増大が伴います。
近年のトレンド「Complex Innovative Design (CID)」

個別化医療(プレシジョンメディシン:遺伝子など個人差に基づき最適治療を選ぶ医療)の進展に伴い、従来の枠組みを超えたデザインが急増しています。
マスタープロトコル:バスケット試験とアンブレラ試験
マスタープロトコルは、1つのプロトコルで複数のサブ試験を行う効率的なデザインです。
がん領域を中心に活用されている試験デザインですが、今後他の領域でも活用が期待されています。
バスケット試験 (Basket Trial)
「1つの薬 × 複数のがん種」。
臓器に関わらず、共通の遺伝子変異を持つ患者を集めて特定の分子標的薬を評価します(同じ分子異常を軸に患者をまとめる考え方)。(参考:日本製薬工業協会 4)
例:同じ遺伝子変異”を持つなら、がんの臓器が違っても同じ薬が効くかを検証したい
- 肺がん
- 大腸がん
- 胆道がん
複数のがん種で同じ遺伝子変異を持つ患者だけを集め、分子標的薬Aを投与して奏効率を評価。
アンブレラ試験 (Umbrella Trial)
「1つのがん種 × 複数の薬」。
特定のがん種(例:肺がん)の中で、遺伝子変異のタイプ別に異なる薬剤を割り付けます(同じがん種内で分子タイプ別に治療を分ける考え方)。(参考:日本製薬工業協会 4)
例:同じ肺がんでも、遺伝子タイプで効く薬は異なるかを検証したい
肺がん患者を検査し、
- 変異X → 薬A
- 変異Y → 薬B
- 変異Z → 薬C
というように同じがん種の中で薬を分けて効果を評価。
プラットフォーム試験
時間経過とともに効果のない治療群を除外し、新しい治療群を追加できる「終わらない試験」構造を持ちます(試験を継続しながら治療群を入れ替えられる仕組み)。
例:新しい治療候補が次々出てくる疾患で、効率よく勝ち残り方式で検証
共通の標準治療を対照にしながら
- 有望な新薬群を追加
- 効果が弱い群は途中で終了
を繰り返す。感染症やがんなどで採用される考え方。
アダプティブデザイン (Adaptive Design)
試験途中のデータ(中間解析:途中時点で行う効果や安全性の評価)に基づいて、事前に計画された手順に沿って試験法を変更するデザインです。
できること: サンプルサイズの再設計、効果が見込めない群の早期中止、優良な群への割付比率の変更などが可能です。
注意点: 恣意的な変更は許されません。タイプIエラー(偽陽性:本当は差がないのに差があると判断する誤り)の増大を防ぐ統計的な調整と、PMDAへの綿密な事前相談が必須です。
例:花粉症の新薬Aが標準薬Bより効くか確認したい
当初の計画では200人で比較する設計。
ただし、中間解析で効果は出ていそうだが予想よりばらつきが大きいことが分かった。
そこで、あらかじめ決めておいた条件に従ってサンプルサイズを300人に増やす。
ベイズ流デザイン (Bayesian Design)
従来の頻度論(P値:偶然による結果の確率を評価する指標)だけにこだわらず、「事後確率(データが得られた後に真である確率)」を用いるアプローチです。
メリット: 過去の試験データなどの「事前情報」を活用できるため、サンプルサイズが限られる希少疾患や小児がん、医療機器開発において効率的です。
例:患者が少ない希少疾患で、無駄なく試験したい
過去の小規模研究やレジストリを事前情報として使い、新しい試験データが入るたびにこの薬が有効である確率を更新。
一定の確率を超えたら早期に有効と判断、低ければ早期中止を検討。
特殊なケース:希少疾患・小規模試験のデザイン
N-of-1試験(単一参加者試験)
一人の患者の中で、実薬とプラセボをランダムに何度も切り替えて評価します。
集団平均ではなく、「その患者個人にとって薬が効くか」を科学的に検証する、究極の個別化医療デザインです。
例:慢性の神経痛で“この患者にとって”本当に効く薬を知りたい
1人の患者に
- 1週間:薬A
- 1週間:プラセボ
- 1週間:薬A
- 1週間:プラセボ
のようにランダムな順で複数回切り替え、痛み日誌や活動量を比較してこの人にAが効くかを判断。
リアルワールドデータ(RWD)と外部対照の活用
疾患登録(レジストリ:患者情報を系統的に集めたデータベース)データなどを対照群(外部対照)として利用し、単群試験の結果と比較する手法です。対照群を置くことが困難な希少疾患での開発を補完します。
例:患者数が少ない希少疾患で新薬Aの有効性を評価したい
新薬Aを使う単群試験を実施したいが、倫理的・人数的に「プラセボ群」を作れない。
そこで、過去に同じ疾患で標準治療を受けた患者のレジストリ(RWD) を集め、外部対照群として比較する。
最適なデザインを選ぶための視点

臨床試験のデザインを選択する際には、以下の複数の判断軸を総合的に評価する必要があります。
どのデザインが「科学的に妥当」「倫理的に許容」「実務的に成立」するかを評価するための観点です。
1. 研究目的(Research Question)に基づく判断軸
臨床試験の中心となる問いは何か?
探索/優越性検証/非劣性検証のどれか
疾患、重症度、治療ライン、バイオマーカー
ORR、PFS、OS、症状改善、バイオマーカー変化など
→ 研究の目的と評価項目が、適切なデザインを選ぶ“根本条件”となる。
2. 疾患と介入特性の判断軸
デザインが成立するための生物学的・臨床的制約は何か?
急性/慢性、進行性か安定性か
可逆的か、持ち越し効果(carryover)が生じるか
→ クロスオーバーが可能か、単群試験が許されるか、対照群が必須かが決まる。
3. 実行可能性(Feasibility)の判断軸
理論上可能でも、実務として成立するか?
→ RCTができる症例数か/小規模デザインが必要かの現実的な判断基盤。
4. 対照群設定の妥当性を評価する判断軸
対照群を置くことが可能か/倫理的か?
→ 対照群の有無は、科学的妥当性と倫理性の両側面から評価される主要判断軸。
5. 同一被験者内比較の適否(クロスオーバー)の判断軸
クロスオーバーが成立する疾患・薬剤か?
→ 用量反応や短期効果を見る場合に有効だが、適応条件が厳しいデザイン。
6. 精密医療(バイオマーカー)適応の判断軸
遺伝子変異や分子サブタイプが試験構造に影響するか?
→ マスタープロトコル採用の可否を決める主要判断軸。
7. 統計的妥当性の判断軸
専門家の視点で科学的成立性を担保できるか?
→ デザインの“科学的成立性”を保証する不可欠の軸。
8. 規制当局(PMDA等)との整合性判断軸
承認戦略・倫理性に照らして受容性があるか?
→ 実際に承認申請を見据える場合は確認すべき軸。
臨床試験デザインに関するよくある質問 (FAQ)
- Qエビデンスレベルが最も高い研究デザインは?
- A
個別の臨床研究では「ランダム化比較試験(RCT)」が最も上位です。さらに複数のRCTを統合・解析した「システマティックレビュー(メタ解析:複数研究を統合して結論の精度を高める方法)」がその上位に位置します。
- QPOC(Proof of Concept)とは医療用語で何ですか?
- A
「概念実証」です。主に第II相試験などで、新薬の効果が動物だけでなくヒトでも期待通りに発揮されるかを確認する重要な段階を指します。
- Q治験の失敗率を下げるためのデザイン上の工夫は?
- A
バイオマーカー(治療効果や病態を反映する測定指標)を活用して効果が出やすい患者層を絞り込むことや、無益な場合に早期中止してリソースを最適化できる「アダプティブデザイン」の導入などが挙げられます。
- QQ. アンブレラ試験とバスケット試験の違いを一言で言うと?
- A
「同じ病気・違う薬=アンブレラ」、「違う病気・同じ薬=バスケット」です。
まとめ
臨床試験デザインは、科学・倫理・統計・規制が複雑に絡み合うパズルです。
近年ではベイズ流やアダプティブデザインなど選択肢が増え、より高度な専門性が求められています。
本記事で得た知識をベースにしつつ、実際の計画段階では必ず統計専門家やPMDAと早期から相談を行うことが、試験を成功に導く最大の近道です。
参考資料・文献一覧
1.PMDA https://www.pmda.go.jp/files/000156372.pdf
2.PMDA https://www.pmda.go.jp/int-activities/int-harmony/ich/0076.html
3.PMDA https://www.pmda.go.jp/files/000269222.pdf
4.日本製薬工業協会 https://www.jpma.or.jp/information/evaluation/symposium/jtrngf0000001cap-att/DS_202302_masterpc.pdf
5.日本製薬工業協会 https://www.jpma.or.jp/information/evaluation/symposium/jtrngf0000001cap-att/DS_202302_hirakawa1.pdf
6.東京大学 https://biostatistics.m.u-tokyo.ac.jp/wp-content/uploads/2023/03/49-206603.pdf